


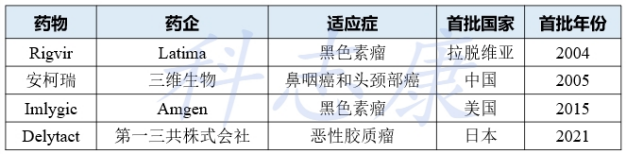
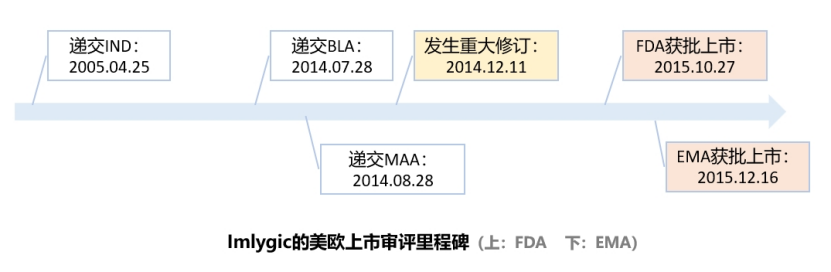
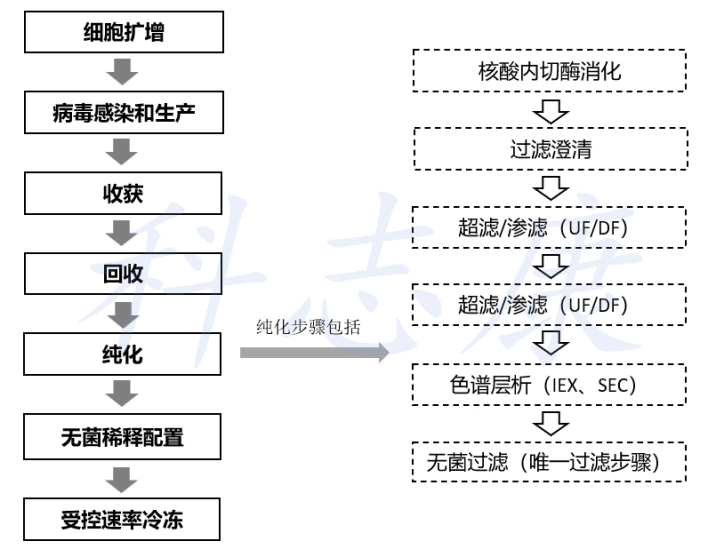
FDA第一个获批上市溶瘤病毒单药Imlygic的CMC解读



END


扫码关注
科志康

关于科志康
上海科志康医药科技有限公司,成立于2016年,是一家以药品监管科学为特色的研发咨询公司,致力于为客户定制法规解决方案,减少和避免在注册申报时“少做、漏做、重做、多做”的风险。
在药品注册时,公司提供高品质的一站式注册申报和模拟现场检查服务,为客户提高药品注册成功率。在研发过程中,公司提供第三方项目管理服务,法规前置,过程管理,协助制药企业提升药品研发效率。
目前,公司已与国内外79个知名制药企业建立合作,取得了100多项IND/NDA/ANDA佳绩。
公司使命:为制药企业提供专家解决方案,提升药品全生命周期合规性
公司愿景:助力制药企业研发和生产,让老百姓吃上放心药




